





GSK.ai Fellow Ron Schwessinger introduces seq2cells – a machine learning framework for single-cell-resolution gene expression prediction directly from long (~200 kb) DNA sequence inputs. seq2cells demonstrates the challenges and value of single-cell gene expression and variant effect prediction, and offers a path to genomic interpretation at uncompromising resolution and scale.
Cells are the fundamental units of life, but we still do not fully understand all 37.2 trillion cells of the human body. Although traditionally classified based on broad morphological and functional characteristics, the advent of single-cell molecular sequencing technologies has allowed exploration of cellular heterogeneity at unprecedented levels. We now rely on the expression patterns of specific genes, which can vary even within seemingly homogeneous cell populations, for identification of distinct cell types. As the wealth of single-cell data has grown, this has revealed previously unknown cellular subtypes, transitional states and rare cell populations that challenge traditional notions of cell type categorization.
We now understand that cells can adopt a continuum of states, driven by unique gene expression profiles and functional behaviours. However, without cellular maps that detail their distinct molecular characteristics and developmental trajectories, we cannot describe their full set of functions and understand the networks that direct their activities. This is an essential part of biomedical research, where it is critical for identification of cell-specific functions, disease mechanisms and potential therapeutic targets. Human genetic variants that impact traits such as disease susceptibility, for example, frequently act through modulation of gene expression in a highly cell-type-specific manner. Computational models capable of predicting gene expression directly from DNA sequence can assist in the interpretation of expression-modulating variants, offering a direct path to predicting the impact of genetic variants on transcript levels as a proxy for cellular function.
Yet predicting gene expression from DNA sequences remains a challenging task for machine learning. As well as capturing complex sequence logic and interactions over large genomic distances, models that aim to predict single-cell gene expression from DNA sequences (known as sequence-to-expression models) must also address challenges such as data sparsity. Although single-cell RNA-sequencing technology offers a particularly powerful approach to tackle heterogenous tissues beyond the resolution of known cell types, its use frequently leads to >90% zero entries in the cell-by-gene expression matrix (a foundational data structure used to identify clusters of cells with similar gene expression profiles), with knock-on effects for analysis. Established sequence-to-expression models only operate using bulk data of broadly defined tissues and cell types [1–4]. However, this limits their ability to characterize heterogeneous tissues and pinpoint cell-type-specific variant effects.
seq2cells: architecture and evaluation
In a recent preprint, we introduce seq2cells – a machine learning framework that captures cell-specific gene expression beyond the resolution of pseudo-bulked data and allows variant effect prediction at single-cell level.
Our framework leverages transfer learning, using models pretrained on bulk-level epigenomic tasks to overcome the context limitations posed by the finite number of genes. Although most DNA language models are currently unable to operate at the sequence context necessary to capture human gene regulation [5–7], we utilized the deep learning model Enformer [4] – which can capture long-range interactions (up to 200 kb away) in the genome – as a pretrained epigenomic base model. Epigenome models such as Enformer are trained on a diverse corpus of epigenomic features that include cell-type-specific CAGE-seq, a technique that maps RNA transcripts roughly to their initiation site and serves as a good proxy for overall RNA transcription levels in different tissues [8–11]. We reasoned that all information required to predict gene expression is therefore encoded in the embeddings of the sequence windows at which CAGE-seq profiles are enriched, predominantly the transcription start site (TSS).
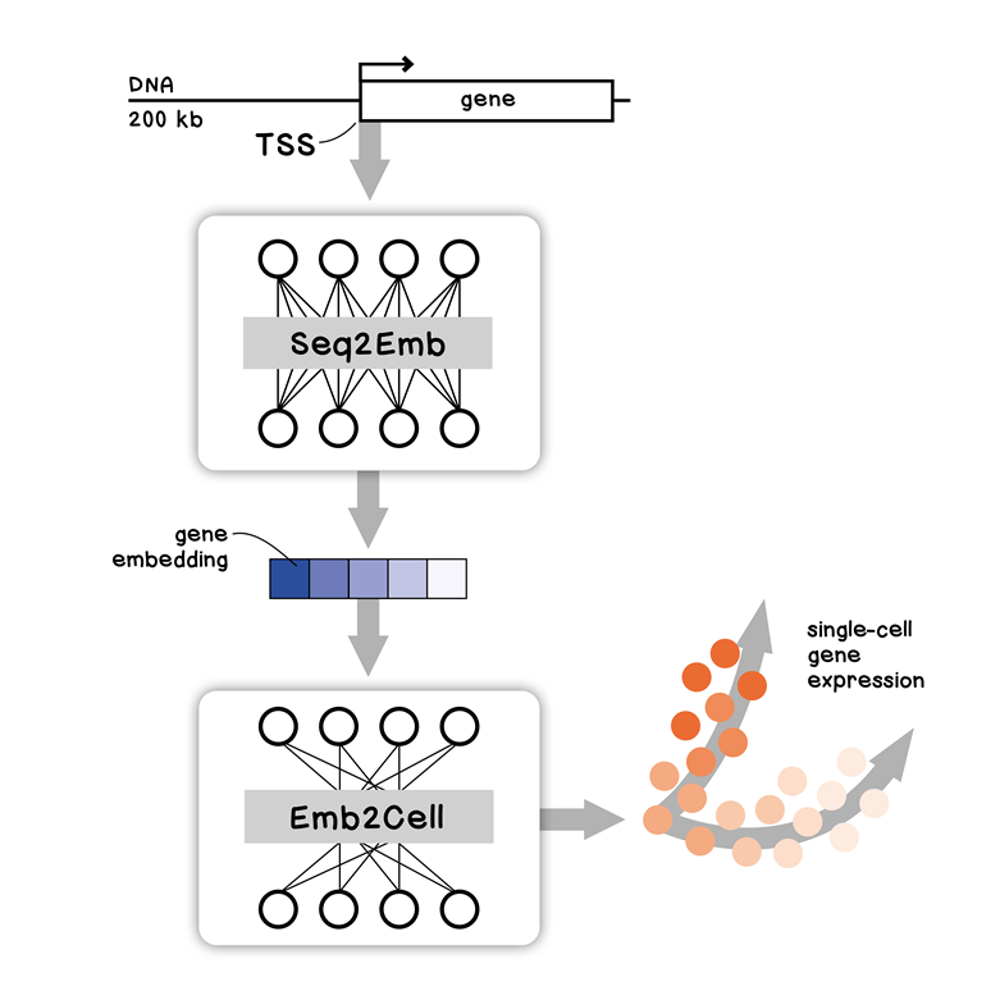
When making a prediction, the model can be run using pre-computed embeddings or a DNA sequence input. This allows us to predict the effect of sequence variants on single-cell expression by comparing the predicted expression of a reference and variant sequence.
Predicting gene expression in single cells
We trained single-cell expression models using a hematopoietic stem cell (HSC) dataset of ∼30,000 cells [12]. Evaluating model performance in predicting cell-specific gene expression on a set of 1,930 genes not seen during training showed the ability of the models to generalize over unseen genes and capture cell-specific gene expression beyond pseudo-bulk resolution.
Evaluating the performance of single-cell expression models, where technical noise and drop-outs obscure the true biological signal, is challenging. We carefully evaluated model performance using raw, artificially denoised and pseudo-bulked data and developed a new strategy to estimate the performance that a perfect model could achieve in this sparse realm. This allowed us to characterize the space between our single-cell model and bulk model performance, leading to the identification of model, sparsity and complexity gaps – each of which could be addressed individually for improvements in future models (see preprint for more information).
Predicting variant effects in single cells
The most direct link between sequence variation and gene expression when attempting to understand disease-linked genetic variability is provided by expression quantitative trait loci (eQTLs) – analyses that are traditionally performed on bulk RNA-sequencing data, often on broad tissues such as whole blood. This makes it difficult to discover variants with cell-type-specific effects unless an affected cell type dominates the bulk data [13]. As variant effects may be highly specific to individual cell types as well as developmental and activation stages, prediction at single-cell resolution offers an opportunity to capture variation within aggregated cell types.
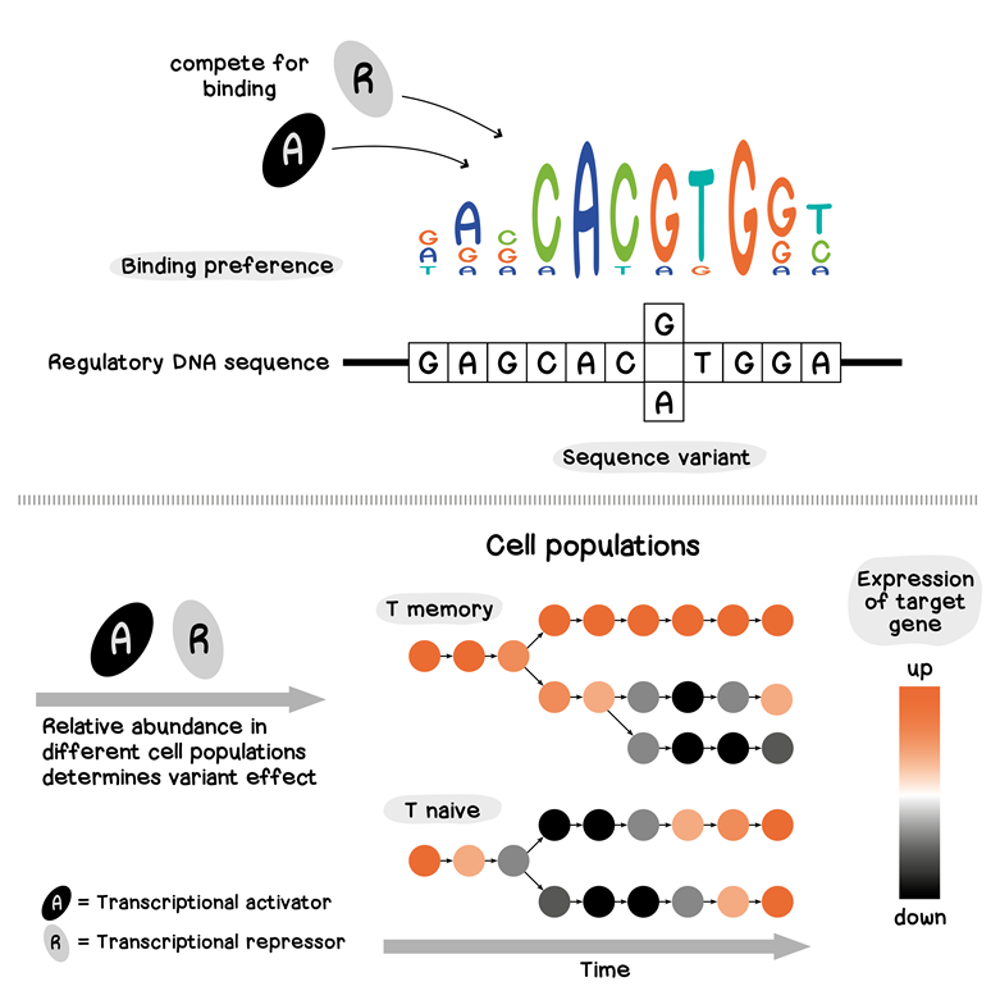
To demonstrate how sequence-based variant effect prediction can characterize cell-specific effects, we predicted the impact of whole-blood eQTLs using our HSC model, calculating the difference between predicted expression of reference and variant alleles for 1,154 variants. Our HSC model predictions suggest that the average effect across cells cannot explain about 80% of eQTL observations. Various reasons may account for this, such as a tissue mismatch, where the affected genes may not be expressed in the cell types investigated or are differently regulated in embryonic or adult cells. However, our models were able to accurately predict whether a genetic variant is linked to an increase or decrease in gene expression, and showed that 43% of variants flip between a positive and negative effect depending on cell type.
Moving beyond averages of many cells, seq2cells allows us to characterize variant effects at greater resolution. This suggests that variant effects can be heterogenous even within an annotated cell type – we found 202 variants (17.5%) exhibit heterogeneity in effect, beyond the resolution of pseudo-bulk data. To gauge if these variations are driven by true biological differences, we characterized the predicted variant effects of exemplary eQTL variants in more detail. This identified bivalent variant effects that can be explained by alterations to DNA binding motifs at sequence level, matched with cell-specific expression of transcription factors (see variant 12_8058101_A_G for an example in our paper). This indicates the presence of multifaceted variant effects that may lead to discrepancies between eQTLs and sequence model predictions, and a highly context-dependent effect on gene expression that can only be fully characterized at single-cell resolution.
Mapping variant effects from T cell activation to T cell development
To evaluate our approach against more complex datasets, we trained seq2cells models on a human T cell development atlas of 250,000 cells and a T cell activation dataset of 650,000 cells [12,14]. The activation dataset, which followed two closely related cell types over a course of time following stimulation, exhibited less cell-to-cell variation compared to the other datasets in this work. Yet despite the reduced variation and increased complexity, the models successfully captured cell-specific gene expression patterns.
To test the ability of our T cell activation and development models to predict single-cell variant effects in these more challenging settings, we utilized pseudo-bulk-based eQTLs, including 480,000 significant variant–gene interactions [14]. Within these data, we found that T cell activation eQTLs also affect developing T cells, suggesting that similar transcriptional programs may be involved in both T cell activation and development. Although some variants showed similar effects throughout both activation and development, others exhibited opposing effects. Further analysis focused on characterizing individual variants across T cell development and activation at single-cell resolution, identifying variants where the predicted effect follows cell type delineation but also variants with predicted effects more granular than the annotated cell types (Fig. 2).
Impact and potential
Here we have demonstrated the utility of seq2cells for single-cell gene expression and variant effect prediction. We successfully used pretrained epigenome models to create DNA sequence embeddings of the TSS that broadly encapsulate transcriptional regulation. This can be transferred to new cell contexts, allowing us to train models that can predict gene expression from sequence determinants that drive gene expression mechanistically (for example, transcription factor motifs), rather than from co-expression patterns of genes [15,16,17].
Our single-cell gene expression models can generalize over unseen genes and capture cell-specific gene expression beyond pseudo-bulk resolution. By demonstrating that single-cell predictions can be flexibly combined into cell types or whole dataset aggregates, we alleviate the need for defining cell types a priori to model training. The results also show that single-cell gene expression prediction enables variant effect prediction at single-cell and single-base-pair resolution, and allows us to pinpoint variant effects to cells rather than cell types – predicting gene expression and variant effects beyond annotated cell clusters. seq2cells shows that predicted effects can be highly cell specific and flip their sign based on the context. This highlights the value of single-cell level variant effect predictions for cell annotation and variant interpretation.
In the future, we hope to aid other gene-level tasks with the information contained in the regulatory DNA sequence of a gene. We believe seq2cells will be a valuable tool for understanding the effect of genetic variation on transcriptional regulation in dynamic cell landscapes, improving our knowledge of cellular diversity and the intricacies of genetic variation in the context of human health and disease.
References
- Zhou, J. et al. Deep learning sequence-based ab initio prediction of variant effects on expression and disease risk. Nature Genetics 2018 50:8 50, 1171–1179 (2018).
- Kelley, D. R. Cross-species regulatory sequence activity prediction. PLOS Computational Biol-ogy 16, e1008050 (2020).
- Agarwal, V. & Shendure, J. Predicting mrna abundance directly from genomic sequence using deep convolutional neural networks. Cell Reports 31, 107663 (2020).
- Žiga Avsec et al. Effective gene expression prediction from sequence by integrating long-range interactions. Nature Methods 2021 18:10 18, 1196–1203 (2021).
- Ji, Y., Zhou, Z., Liu, H. & Davuluri, R. V. Dnabert: pre-trained bidirectional encoder representations from transformers model for dna-language in genome. Bioinformatics 37, 2112–2120 (2021).
- Trotter, M. V., Nguyen, C. Q., Young, S., Woodruff, R. T. & Branson, K. M. Epigenomic language models powered by cerebras. arXiv (2021).
- Dalla-Torre, H. et al. The nucleotide transformer: Building and evaluating robust foundation models for human genomics. bioRxiv (2023).
- Kawaji, H. et al. Comparison of cage and rna-seq transcriptome profiling using clonally amplified and single-molecule next-generation sequencing. Genome Research 24, 708–717 (2014).
- Karollus, A., Mauermeier, T. & Gagneur, J. Current sequence-based models capture gene expression determinants in promoters but mostly ignore distal enhancers. bioRxiv (2022).
- Huang, C. et al. Personal transcriptome variation is poorly explained by current genomic deep learning models. bioRxiv (2023).
- Sasse, A. et al. How far are we from personalized gene expression prediction using sequence-to-expression deep neural networks? bioRxiv (2023).
- Park, J. E. et al. A cell atlas of human thymic development defines t cell repertoire formation. Science 367 (2020).
- Umans, B. D., Battle, A. & Gilad, Y. Where are the disease-associated eqtls? Trends in Genetics 37, 109–124 (2021).
- Soskic, B. et al. Immune disease risk variants regulate gene expression dynamics during cd4+ t cell activation. Nature Genetics 2022 54:6 54, 817–826 (2022).
- Cui, H. et al. scgpt: Towards building a foundation model for single-cell multi-omics using generative ai. bioRxiv (2023).
- Theodoris, C. V. et al. Transfer learning enables predictions in network biology. Nature 2023 618:7965 618, 616–624 (2023).
- Hao, M. et al. Large scale foundation model on single-cell transcriptomics. bioRxiv 2023.05.29.542705 (2023).

